LOTUS DOCUMENTATION
Index
Installation || Tutorials|| Using LotuS from the commandline|| Mapping file configuration|| LotuS output|| Configuring sdm quality filter|| Frequently asked questions||Installation
DOWNLOADS
The installation of LotuS requires lotus.pl, sdm, sdm configuration file, LotuS configuration file. These can be downloaded from here.
To install, run "tar -xzf lotus_*.tar.gz" in the folder you want to install LotuS to.
Due to license restrictions, usearch
has to be downloaded and installed by the user,
all other programs are automatically installed and configured by executing the autoInstall.pl script that is in the downloaded folder.
All proprietary software used in your specific LotuS run will be listed, including the relevant citation, in "output_dir/citations.txt". Please refer to the SILVA Terms of Use/License Information.
LOTUS CONFIGURATION
If the autoInstaller script was used, only the path to usearch needs to be configured:
Go to the LotuS folder on your harddrive and open lotus.cfg, the LotuS configuration file. In this file, the paths of the downloaded software has to be set to the program locations on your local machine.
Search for a line starting with "usearch" and enter the absolute path to the usearch executable after a space character, e.g.:
usearch /Users/Tomas/bin/usearch/usearch7.0.1001_i86linux32LotuS is now ready to be used.
Important for MAC users:In some cases sdm will not compile correctly, but this is required for the pipeline to run. If this happens, please make sure that your system supports C++11 (with the programs gcc / clang)
Any optional step within the pipeline can be deactivated by making the path to the software invalid within the lOTUs.cfg file (e.g. by deleting a single chracter in the absolute path).
UPDATING LOTUS
LotuS can be easily updated using the autoinstaller.pl script, if LotuS was installed using the autoinstaller and the path to a working usearch is set. Now, runnning the autoinstaller again ("./autoInstall.pl") will query the user if new updates should be installed.
TUTORIALS
The following tutorials go through the usage of LotuS on an example dataset and the analysis of LotuS output files in the R statistical language:- LotuS tutorial with two 454 input files
- Making sense of taxa level tables in R using numerical ecology
LotuS command line tool
LotuS is a perl script that links several programs to demultiplex and cluster 16S amplicon sequences. The general syntax to LotuS is:
Perl lotus.pl –i (data) –o (results folder) –m (mapping file) –s (sdm_454.txt)
- -i [input fasta / fastq / dir]. In case that fastqFile or fnaFile and qualFile were specified in the mapping file, this has to be the directory with input files
- -o [output dir] Warning: The output directory will be completely removed at the beginning of the LotuS run. Please ensure this is a new directory or contains only disposable data!
- -m [mapping file]
- -barcode [file path to fastq formated file with barcodes (this is a processed mi/hiSeq format). The complementary option in a mapping file would be the column "MIDfqFile".]
- -q/-qual [input qual file (not defined in case of fastq or input directory)]
- -s/-sdmopt [sdm option file, defaults to "sdm_454.txt" in current dir]
- -c/-config [LotuS.cfg, config file with program paths]
- -t/-tmpDir [temporary directory used to save intermediate results]
- -keepTmpFiles [1: save extra tmp files like chimeric OTUs or the raw blast output in extra dir; 0: don't save these, default 1]
- -highmem [1: highmem mode which has much faster execution speed but can require substantial amounts of ram (e.g. hiSeq: ~40GB).0 deactivates this, reducing memory requirements to < 4 GB, default=1]
- -thr/-threads [number of threads to be used]
- -p/-platform [sequencing platform: PacBio, 454, miSeq or hiSeq]
- -flash_param [custom flash parameters, since this contains spaces the command needs to be in parentheses: e.g. -flash_param "-r 150 -s 20". Note that this option completely replaces the default -m and -M flash options (i.e. need to be reinserted, if wanted)]
- -custContamCheckDB [This option checks in analogy to the phiX filter step in a custom DB (e.g. mouse genome, needs to be fasta format), for contaminant OTUs that are more likely to derrive from this genome than e.g. bacteria. Example: "-custContamCheckDB /YY/mouse.fna". Default: "". ]
- -saveDemultiplex [1: Saves all demultiplexed reads (unfiltered) in the [outputdir]/demultiplexed folder, for easier data upload. 2: Only saves quality filtered demultiplexed reads and continues LotuS run subsequently. 3: Saves demultiplexed file into a single fq, saving sample ID in fastq/a header. 0: No demultiplexed reads are saved. Default: 0]
- -tolerateCorruptFq [1: continue reading fastq files, even if single entries are incomplete (e.g. half of qual values missing). 0: Abort lotus run, if fastq file is corrupt. Default 1]
- -itsextraction [1: use ITSx to only retain OTUs fitting to ITS1/ITS2 hmm models; 0: deactivate; Default=1]
- -itsx_partial [0-100: parameters for ITSx to extract partial (%) ITS regions as well; 0: deactivate; Default=0]
- -CL/-clustering [(1) use UPARSE, (0) use otupipe (deprecated), (2) use swarm and (3) use cd-hit, default 1]
- -id [clustering threshold for OTU's, default 0.97]
- -swarm_distance [clustering threshold for OTU's when using swarm clustering, default 1]
- -derepMin [minimum size of dereplicated clustered, one form of noise removal. Can also have a more complex syntax, see examples. Default 1]
- -chim_skew [skew in chimeric fragment abundance (uchime option), default 2]
- -readOverlap [the maximum number of basepairs that two reads are overlapping, default 300]
- -deactivateChimeraCheck [0: do OTU chimera checks. 1: no chimera Check at all. 2: Deactivate deNovo chimera check. 3: Deactivate ref based chimera check.Default = 0]
- -xtalk [(1) check for crosstalk; note that this requires in most cases 64bit usearch, default 0]
- -rdp_thr [Confidence threshold for RDP, default 0.8]
- -utax_thr [Confidence thresshold for UTAX, default 0.8]
- -LCA_frac [min fraction of reads with identical taxonomy, default 0.9]
- -TaxOnly [skip most of the lotus pipeline and only run a taxonomic classification on a fasta file, provided via "-i" (could be an OTU fasta)]
- -keepUnclassified [1 includes unclassified OTUs (i.e. no match in RDP/Blast database) in OTU and taxa abundance matrix calculations; 0 does not take these OTU's into account, default:1]
- -simBasedTaxo [0: deavtivated (default RDP used instead); 1 or blast: use Blast; 2 or lambda: use LAMBDA to search against a 16S reference database for taxonomic profiling of OTUs; 3 or utax]: use UTAX with custom databases. Default 0]
- -refDB The following keywords (in combination) are possible:
[SLV: Silva LSU (23/28S) or SSU (16/18S), GG: greengenes (only 16S available), UNITE (ITS focused on fungi), PR2 (SSU focused on Protists), beetax (bee gut specific database and tax names) or HITdb (a database specialized only on human gut microbiota). Decide which reference DB will be used for a similarity based taxonomy annotation, default is "SLV".
Databases can be combined, with the first having the highest prioirty. E.g. PR2,SLV would first use PR2 to assign OTUs and all unaasigned OTUs would be searched for with SILVA, given that -amplicon_type LSU was set.
Can also be a custom fasta formatted database: in this case provide the path to the fasta file as well as the path to the taxonomy for the sequences using -tax4refDB. See online help on how to create a custom DB.] - -tax4refDB [in conjunction with a custom fasta file provided to argument -refDB, this file contains for each fasta entry in the reference DB a taxonomic annotation string, with the same number of taxonomic levels for each, tab separated. See example]
- -amplicon_type [LSU: large subunit (23S/28S) or
SSU: small subunit (16S/18S). Default SSU] or
ITS: internal transcribed spacer, only supported for fungal data using UINTE database - -tax_group [bacteria: specialized 16S rDNA annnotation, fungi: fungal 18S/23S/ITS annotation. Default "bacteria"]
- -useBestBlastHitOnly [1: don't use LCA (last common ancestor) to determine most likely taxnomic level (not recommended), 0: (default) LCA algorithm]
- -redoTaxOnly [1: only redo the taxonomic assignments (useful for replacing a DB used on a finished lotus run), 0: normal lotus run, default]
- -greengenesSpecies [1: Create greengenes output labels instead of OTU (to be used with greengenes specific programs such as BugBase). Default: 0]
- -pseudoRefOTUcalling [1: create Reference based (open) OTUs, where the chosen reference database (SLV,GG etc) is being used as cluster center. Default: 0]
MAPPING FILE CONFIGURATION
The mapping file, specified via the "-map" or "-m" argument to LotuS, is used within the pipeline to demultiplex fasta + qual or fastq files, by using the program sdm (included in LotuS). The first line in the mapping file is the header and has to start with "#". Entries have to be tab separated and the mapping file should be stored as text file. The number and names of columns are not limited (this will be exported to the .biom file) and only columns with column names in the table below will be used for processing the read files. Thus, the desired subparts of the pipeline are activated by having these column names in the header of the mapping file (see also Usage examples)."automap.pl" creates automatically LotuS compatible maps from input dirs, if the run is already demultiplexed. Use perl automapl.pl to get instructions on how to use this helper script.
| Column name | Function |
| SampleID | Sample Identifier, has to be unique for each Barcode. |
| BarcodeSequence | The Barcode (MID) tag assigned to each sample. |
| Barcode2ndPair | in case of dual indexed reads, use this column to specify the BC on the 2nd read pair |
| ForwardPrimer | Sequence used for 16S amplification, usually (unless paired end mode) is after the Barcode. Can contain IUPAC redundant nucleotides. The old tag LinkerPrimerSequence also works for this option. |
| ReversePrimer | Reverse Primer Sequence (IUPAC code) |
| fastqFile | Gives relative location of fastq file, such that [-i][fastqFile] gives the absolute path to fastq file |
| fnaFile | see fastqFile. However, fasta formated file instead of fastq format |
| qualFile | see fastqFile. However, quality file corresponding to fasta file instead of fastq format |
| SampleIDinHead | ID in header of fasta/fastq file, that identifies Sample //This replaces Barcode (MID) scanning\\ |
| MIDfqFile | extra fastq file that contains only MIDs (equivalent to LotuS script option "-barcode"). Requires paired reads. |
| CombineSamples | Option to combine samples, that may be distributed across several files. A new tag is set here and all samples with this specific tag will be merged into a new tag with tag as SampleID. Note that SampleID's themselves have still to be unique. |
If more than 1 sequencing run was used, it is often easier to demultiplex all fasta / fastq sequences at once. For this purpose LotuS/sdm offers either the fastqFile column, or in case of fasta+qual input files, the two columns fnaFile and qualFile. Within these columns the relative path towards input files can be specified.
Note that this format is very similar to the Qiime mapping file format, but for additional options exclusive to LotuS.
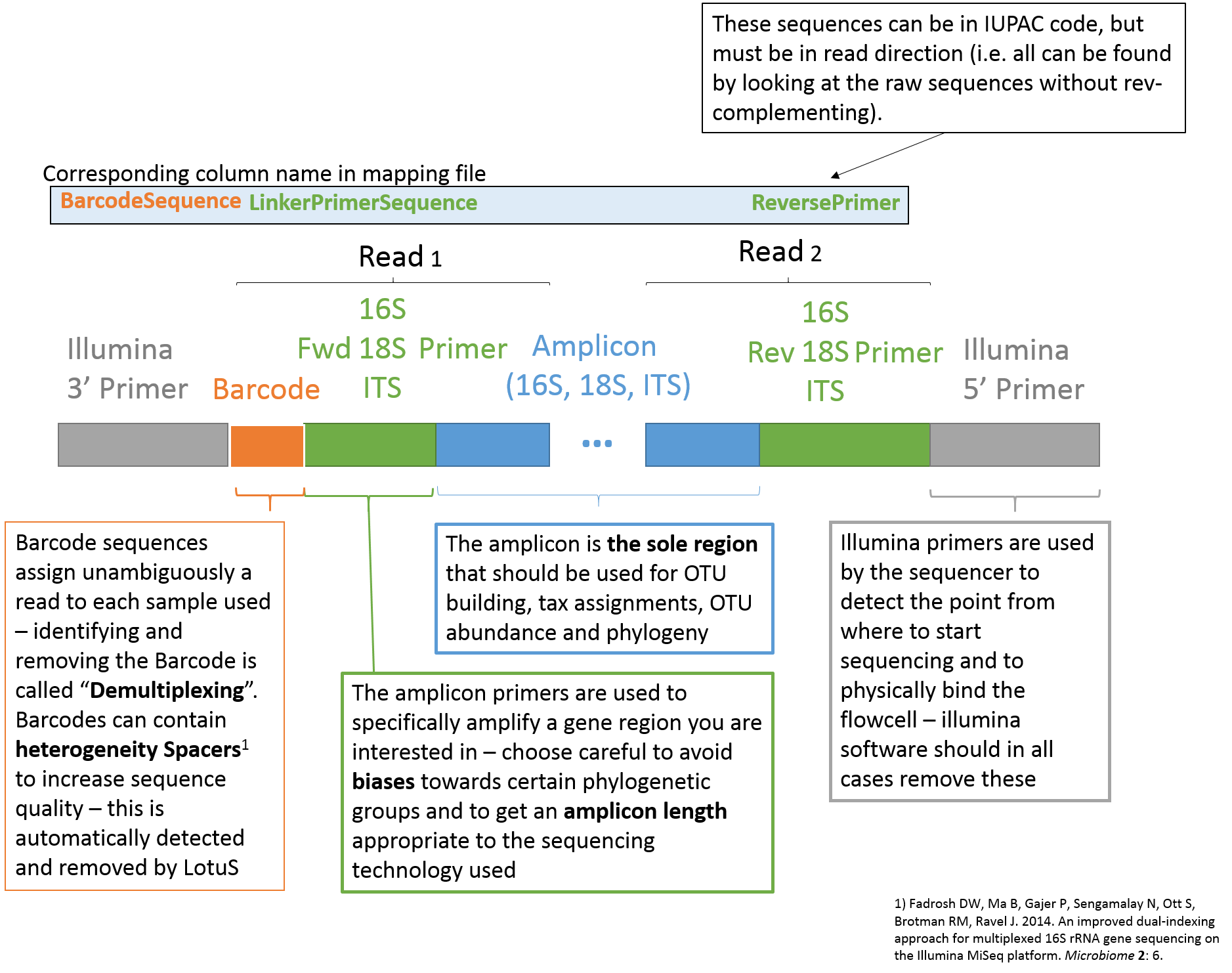 |
LOTUS OUTPUT
After the LotuS run has finished, the output folder specified via the -o option contains the following files and subfolders:| file/folder | Function |
| /LotuSLogS/ | Directory with log files for OTUpipe/UPARSE |
| /LotuSLogS/demulti.log.* | Log File for sdm (includes a log file for each single fna/fastq file) |
| /LotuSLogS/demulti.acceptsPerFile.log | Number and percentage of reads that was accepted per sample - can be used to find samples that consistently had reduced read quality. |
| /LotuSLogS/demulti.acceptsPerSample | Percentage of reads that was accepted per input file - can be used to find sequencer output file that consistently had reduced read quality. |
| /LotuSLogS/demulti_lenHist.txt | Length histogram of sequences that passed sdm |
| /LotuSLogS/demulti_qualHist.txt | Quality histogram of sequences that passed sdm |
| /LotuSLogS/SeedExtensionStats.txt | Min/Avg/Max stats on Seed sequence length, quality accumulated error and similarity to OTU consensus sequence |
| /LotuSLogS/LotuS_runlog.txt | This file tracks overall progression and if certain steps failed. |
| /primary/ | Directory with copies of sdm options and mapping file |
| /higherLvl/ | Directory with Species, Genus, Family, Class, Order and Phylum abundance matrices |
| hiera_BLAST.txt | OTU taxonomy assignments based on Blastn |
| hiera_RDP.txt | OTU taxonomy assignments based on RDP classifier |
| OTU.txt | OTU abundance matrix |
| OTU.biom | biom formatted OTU abundance matrix |
| otuMultAlign.fna | Multiple Alignment of OTU’s |
| otus.fa | Fasta formatted extended OTU seed sequences |
| Tree.tre | Newick formatted phylogenetic tree between sequences |
SDM OPTION FILE CONFIGURATION
This file contains options for the quality filtering of sequences, each option is described again in a comment above the relevant flag (comments can be written in this configuration file by starting the line with "#"). Some options start with a "*". These are relaxed parameters (i.e. they have to be less restrictive) for the secondary quality filter. Sequences in this mid-quality range will not be used for the OTU building, but only mapped to the OTU's.| Flag | Function |
| minSeqLength | minimal required sequence length AFTER removal of Primers, Barcodes and trimming. |
| maxSeqLength | maximum sequence length |
| TruncateSequenceLength | runcate total Sequence length to X (length after Barcode, Adapter and Primer removals), set to -1 to deactivate. |
| minAvgQuality | minimal required average quality AFTER removal of Primers, Barcodes and trimming. |
| maxAccumulatedError | Probabilistic max number of accumulated sequencing errors. After this length, the rest of the sequence will be deleted. Complimentary to TrimWindowThreshhold. (-1) deactivates this option. |
| BinErrorModelMaxExpError, BinErrorModelAlpha | Binomial error model of expected errors per sequence (see Arxiv paper). (BinErrorModelAlpha -1) deactivates this option. |
| maxAmbiguousNT | maximum ambiguous bases in Sequence |
| maxHomonucleotide | maximum homonucleotide run length |
| QualWindowWidth, QualWindowThreshhold | Filter whole sequence if one window of quality scores is below QualWindowThreshhold |
| TrimWindowWidth, TrimWindowThreshhold | Trim the end of a sequence if a window falls below quality threshhold. Useful for removing low qulaity trailing ends of sequence |
| keepBarcodeSeq, keepPrimerSeq | keep Barcode / Primer Sequence in the output fasta file - in a normal 16S analysis this should be deactivated (0) for Barcode and deactivated (0) for primer |
| fastqVersion | set fastqVersion to 1 if you use Sanger, Illumina 1.8+ or NCBI SRA files. Set fastqVersion to 2, if you use Illumina 1.3+ - 1.7+ or Solexa fastq files. |
| TechnicalAdapter | if one or more files have a technical adapter still included (e.g. TCAG 454) this can be removed by setting this option |
| TrimStartNTs | delete X NTs (e.g. if the first 5 bases are known to have strange biases) |
| PEheaderPairFmt | correct PE header format (1/2) this is to accomodate the illumina miSeq paired end annotations 2="@XXX 1:0:4" instead of 1="@XXX/1". Note that the format will be automatically detected |
| AmpliconShortPE | This option should be "T" if your amplicons are possibly shorter than a read in a paired end sequencing run (e.g. amplicon of 300 in 250x2 miSeq is "T"). This works in conjunction with keepPrimerSeq (should be "F") and a the ReversePrimer field in the mapping file. |
| RejectSeqWithoutRevPrim, RejectSeqWithoutFwdPrim | sets if sequences without a forward (LinkerPrimerSequence) primer will be accepted (T=reject ; F=accept all); default=F |
| CheckForMixedPairs | this option should be "T" if your run possibly swaps fwd and rev pairs (this can happen depending on how your sequence provider was running illumina). |
| CheckForReversedSeqs | this option should be "T" if youwant to double check, if sometimes reads are completely reverse-translated. |
FAQ
Q: Is there a windows version?A: No. The main problem is here that some of the properitary software used in LotuS is not Windows compatible. Therefore we will not release a Windows version any time soon.
Q: I need to upload my demultiplexed data to a public repository, how do I get the demultiplexed files?
A: Use the option "-saveDemultiplex 1".
Q: What is the difference between sdm and LotuS?
A: sdm is an integral part of LotuS, responsible for quality filtering, demultiplexing, sequence format changes and seed extension. However, sdm was conceptualized as a stand-alone software. E.g. I personally use sdm to quality filter sequence files before assembling bacterial genomes. To get more information of the sdm interface, execute the sdm binary without arguments ("./sdm") and a help is displayed.
Q: Can I use gzip compressed files?
A: Yes. For config and mapping files this is not supported but all sequence files can be compressed using gzip. Just make sure the file ending is ".gz" and sdm will assume this is a compressed file. Note that on some systems sdm compilation with zlib library may fail; the autoinstaller attempts to detect this and compile sdm without zlib support.
Q: What part of the sequence is cut?
A: Everything that is the remainder of technical processes is removed, if possible. E.g. Giving Barcodes in the sequence, will remove all sequence upstream of the Barcodes (including heterogenity spacer, illumina primer). If Fwd and Rev 16S amplification Primers are provided in the mapping file (and they are found in a read), everything upstream of these is removed (including Barcodes, het spacer etc.).
Q: Should I keep unclassified OTUs (-keepUnclassified option)?
A: In general we do not recommend this, as these sequences could be environmental sequences that are not 16S (e.g. eukaryotic genomes contain regions with distant homology to frequently used 16S primer pairs). If you assume that you might have new phyla in your sample or species very distant from known organisms, you can deactivate this option, but I would still recommend to cross check with e.g. NCBI Blast that an unknown OTU is not a random gene. This option is activated by default, because it was confusing if a large part of reads went silently missing.
Q: My Barcodes are reverse complemented, can I set an option to take care of this?
A: This should be automatically detected: sdm has an inbuilt algorithm that checks in the first 5000 sequences, if more reverse complemented BCs can be detected and will use this information for the rest of the file. However, BCs have to be consistent in their direction, as the direction information is assummed to be the same within each file.
Q: I do not want to use RDP assigned taxonomies, but use reference databases. Should I use the SILVA or Greengenes 16S ref databases?
A: Both databases have a large selection of taxa included, though SILVA has a faster release cycle and is currently more up to date, the last GG release was in 2013. Also, Silva includes LSU and Eukaryotic (18S/28S) sequences, so greengenes can only be used for bacterial SSU (16S) sequences.
Q: How to choose a good cutoff length of sequences?
A: Changing the TruncateSequenceLength and minSeqLength is fine tuning to your data set - just remember to keep these parameters equal. As a general rule of thumb: you want to have as long as possible reads, but every read below that length will be excluded. Further, the accumulated error has to be below e.g. 0.5 (parameter maxAccumulatedError), so longer reads means more errors and here you have to fine a good balance between read errors and sequence length.
(All parameters are in the sdm_XXX.txt option files)
Q: How to further optimize my LotuS run?
A: First of all you need to optimize the number of sequences you gain vs the number of errors you allow to pass into OTU building. This is mainly done in the sdm_opt* files, the files I provide on the website are just general purpose suggestions.
Second, choose your clustering algorithm according to your needs. UPARSE is my general recommendation; some users have reported better clusterings in the usearch7x versions. SEED clusters are very sensitive, to a point where read errors could cluster into a new OTU, but if you need pseudo-strain resolution this might work for you. cd-hit clusters are very uniform, that is no dynamic adaptation of identity deprending on cluster size/shape like uparse and swarm do. These are plain "good old clusters".
Third, think about what taxonomic assignments you need and from which database.
RDP provides often a very robust assignment at genus level, but greengenes/Silva can allow annotations at species level.
Q: I'm testing over a set of our samples and wondering why, no matter if using GG or SLV database, never a single OTU is classified at species level? For the records, with QIIME I get at least some OTU assigned at species level.
Depends on the environment you work in. So for gut environment, you should get a good fraction of OTUs assigned to species level; other environments like Arctic samples are often not well represented at species level.
In LotuS we avoid the best-hit-assignment (unless specified with option -useBestBlastHitOnly). LotuS has a least common ancestor algorithm that looks if there are several hits of similar quality to different species of the same genus/family/class etc. It then goes to the node of hits that capture 95% of hits (with some additional checks if reference sequences even have a species assignment etc). Further, if the identity of the hit is below a certain threshold (set in the lotus.cfg file), it will not assign species, genus, family etc. labels, if not higher than e.g. 95% similarity to the database hit. This is to prevent falsely assigned species names, even if this means retuning a lot of genera without species assignments.
Q: Where are reads exactly removed during the LotuS run
A: 1st) during Quality filtering and also dereplication (these are later counted into the OTU matrix by similarity comparison, but not used for OTU construction). 2nd) is during Chimera detection steps. 3rd) (optional) unassigned OTU's and all associated reads can be removed (option -keepUnclassified 0).
Q: How can I cite LotuS in my work?
A: The citation is Hildebrand F, Tadeo RY, Voigt AY, Bork P, Raes J. 2014. LotuS: an efficient and user-friendly OTU processing pipeline. Microbiome 2: 30. and the paper is available here.